Classic PKAN: Understanding Early Onset Symptoms and Progression
Pantothenate Kinase-Associated Neurodegeneration, commonly known as PKAN disease, represents one of the most prevalent forms of Neurodegeneration with Brain Iron Accumulation (NBIA) disorders. Affecting approximately 30-35% of the NBIA population, PKAN is a complex genetic condition characterized by progressive neurological decline. While a spectrum of symptoms and progression rates exists, leading to classifications like classic and atypical forms, this article will delve specifically into classic PKAN, focusing on its early onset, distinctive symptoms, and rapid progression. Understanding the nuances of classic PKAN is crucial for early diagnosis, proactive management, and improving the quality of life for affected individuals and their families.
The Genetic Underpinnings of PKAN
At its core, PKAN disease is caused by mutations in the PANK2 gene, located on chromosome 20. This gene carries the vital instructions for producing an enzyme called pantothenate kinase. Pantothenate kinase plays a critical role in Coenzyme A (CoA) biosynthesis, a fundamental metabolic pathway. When mutations in the PANK2 gene occur, the production of this enzyme is impaired or completely absent. The precise mechanism by which this deficiency leads to nerve cell damage and the characteristic iron buildup in specific regions of the brain remains an active area of research. Scientists are working diligently to unravel these complexities, hoping to unlock new therapeutic avenues.
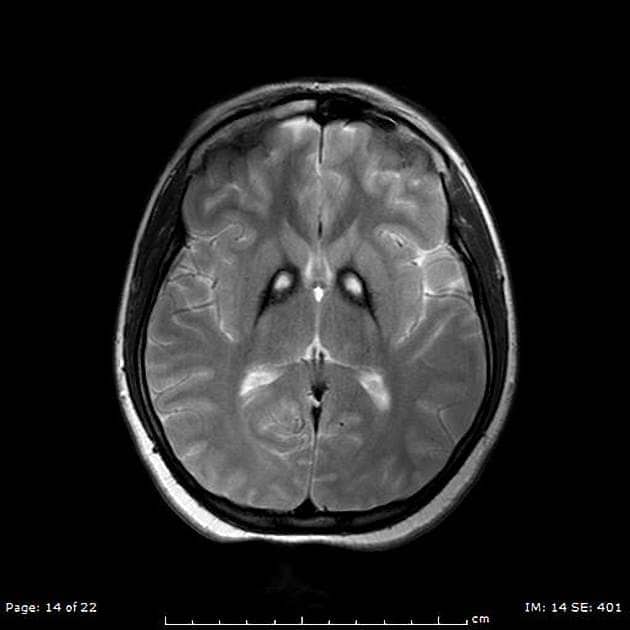
The absence or dysfunction of pantothenate kinase is thought to disrupt various cellular processes, particularly those crucial for brain health. The subsequent accumulation of iron in the globus pallidus and substantia nigra—deep brain structures involved in movement control—is a hallmark feature visible on MRI scans. While the iron itself is not believed to be the primary cause of neurodegeneration, it is a significant indicator of the underlying pathological processes at play.
Early Onset and Rapid Progression in Classic PKAN
Individuals diagnosed with classic PKAN typically experience a more aggressive and rapidly progressive course of the disease compared to those with the atypical form. A defining characteristic of classic PKAN is its early onset. According to published literature, symptoms usually manifest before the age of six in about 90% of patients. While the average age of onset is around 3 years and 4 months, symptoms can appear as early as 6 months or as late as 12 years. This broad range underscores the importance of vigilance in young children presenting with unexplained neurological symptoms.
The initial signs of classic PKAN can often be subtle, making early diagnosis challenging. Parents might first notice that their child seems "clumsy" or struggles with motor skills that peers are acquiring more easily. This early clumsiness gradually evolves into more noticeable problems with walking, balance, and coordination. As the disease progresses, falling becomes increasingly frequent, posing a significant risk of injury. Because children with classic PKAN often have difficulty protecting themselves during falls, they are prone to repeated injuries, particularly to the face and chin.
The rapid progression of classic PKAN means that functional decline can be swift. Many individuals with the classic form require a wheelchair by their mid-teens, losing the ability to move or walk independently within 10 to 15 years after the initial appearance of symptoms. This significant loss of mobility profoundly impacts daily life and necessitates comprehensive support and adaptive strategies.
Unfolding Symptoms: A Deeper Look at Classic PKAN Manifestations
The symptom profile of classic PKAN is diverse, primarily affecting motor function, speech, and, in some cases, vision. Understanding these specific manifestations is key to timely intervention and symptom management.
Dystonia: A Pervasive Early Symptom
One of the most debilitating and consistently present symptoms in classic PKAN is dystonia. This movement disorder causes involuntary muscle contractions and spasms, leading to repetitive or twisting movements and abnormal postures. Dystonia often emerges early in the disease progression and can affect various parts of the body, though cranial and limb dystonia are particularly frequent. Cranial dystonia can impact the muscles of the head, neck, eyes, tongue, mouth, and throat, while limb dystonia affects the arms and legs.
The severe dystonia seen in classic PKAN can lead to significant complications. For instance, tongue dystonia can cause recurrent self-trauma, such as tongue biting, further exacerbated by impacts during falls. Managing dystonia is a cornerstone of care. Botulinum toxin injections are often a first-line treatment for localized dystonia, effectively reducing muscle spasms and pain. In extreme cases of oral dystonia, the difficult decision to remove all teeth may be necessary to prevent severe injury and facilitate feeding. Beyond direct dystonia, the combination of extreme bone stress from involuntary muscle contractions and osteopenia (reduced bone density) increases the risk of bone fractures, highlighting the need for vigilance and bone health monitoring.
For more detailed information on managing this challenging symptom, you can explore resources like PKAN & Dystonia: Navigating Involuntary Muscle Contractions.
Challenges with Speech and Swallowing
As classic PKAN progresses, individuals often develop dysarthria, a speech disorder characterized by difficulty using or controlling the muscles involved in speech production (mouth, tongue, larynx, vocal cords). This can manifest in various ways, making speech difficult to understand: stuttering, slurring, or speech that is soft, hoarse, or raspy. Speech therapy plays a vital role in helping individuals maintain communication abilities for as long as possible and finding alternative communication methods as needed.
Chewing and swallowing difficulties (dysphagia) are also common and can become severe enough to necessitate the use of a feeding tube (gastrostomy tube) to ensure adequate nutrition and hydration. This is often a direct consequence of oral and pharyngeal dystonia. Poor nutrition and aspiration due to swallowing problems are serious secondary effects that can significantly impact health and life expectancy.
Other Neurological and Physical Manifestations
- Corticospinal Tract Involvement: The communication pathways between the brain and limbs are affected, leading to spasticity (stiff, tight muscles) and hyperreflexia (overactive reflexes). These contribute to the overall motor dysfunction and difficulty with movement.
- Retinal Degeneration: Deterioration of the retina at the back of the eye can occur, potentially impacting vision. Regular ophthalmological evaluations are important to monitor and address any visual impairments.
Managing Classic PKAN and Looking to the Future
While there is currently no cure for PKAN disease, management focuses on alleviating symptoms, improving quality of life, and preventing complications. A multidisciplinary approach is essential, involving neurologists, physical therapists, occupational therapists, speech-language pathologists, nutritionists, and social workers. Early diagnosis and intervention are critical to proactively manage symptoms and support the child's development.
Practical tips for families and caregivers include:
- Regular Therapy: Consistent physical, occupational, and speech therapy can help maintain mobility, improve communication, and adapt to increasing challenges.
- Nutritional Support: Close monitoring of weight and nutrient intake is crucial. Dietary modifications, thickened liquids, and eventually feeding tubes, can prevent malnutrition and aspiration.
- Dystonia Management: Alongside botulinum toxin, medications and sometimes deep brain stimulation (DBS) can be considered to manage severe dystonia.
- Safety Measures: Adapting the home environment to prevent falls, using protective gear, and ensuring wheelchair accessibility are paramount.
- Emotional and Social Support: Connecting with support groups and mental health professionals can help individuals and families cope with the emotional toll of a progressive disease.
The progression of classic PKAN places individuals at a higher risk of premature death, often due to secondary complications such as severe swallowing difficulties leading to malnutrition or aspiration pneumonia, or respiratory issues from severe dystonia. However, advancements in medical care, particularly in managing these secondary effects, mean that more individuals with PKAN are now living into adulthood. Ongoing research into the genetic and biochemical pathways of PKAN Disease: Exploring Symptoms, Genetic Causes, and Progression continues to offer hope for future disease-modifying therapies.
Conclusion
Classic PKAN is a challenging neurodegenerative disorder characterized by its early onset, rapid progression, and a complex array of motor, speech, and other neurological symptoms. While the journey with classic PKAN can be difficult, a comprehensive and proactive approach to care, involving a multidisciplinary team and strong family support, can significantly improve the quality of life for affected individuals. Continued research, alongside improvements in symptomatic management, holds the promise of better understanding and, ultimately, more effective treatments for this rare and impactful condition.